1000 Genomes Project
 |
| Image source: Wikipedia |
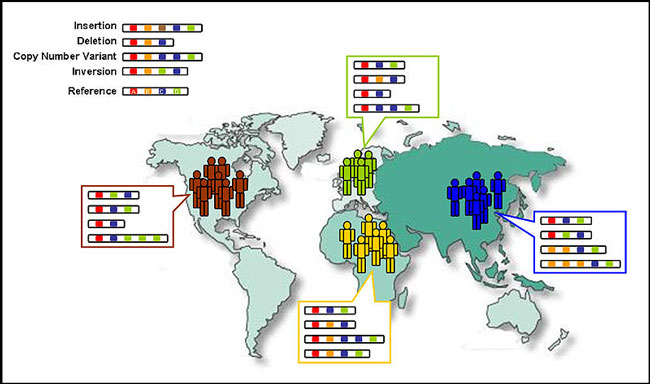
The overall goal of the 1000 Genomes Project is the generation of a nearly complete catalog of common human genetic variants (defined as having a frequency of 1% or higher). This catalog will include Single Nucleotide Polymorphisms (SNPs), copy number variants (CNVs), and short insertion and deletion polymorphisms (Indels) and will be generated by high-quality sequence data obtained from a geographically diverse collection of individuals. These data will be critical to genetic association studies of complex diseases such as cardiovascular disease, various cancers and neurological diseases such a schizophrenia and autism. The project is international in scope with researchers and funding contributions from the US, UK, China and Germany.
The 1000 Genomes Project will also offer insight into the performance of so-called “next generation sequencing” platforms. These technologies offer both substantially increased throughput and reduced cost when compared to traditional Sanger dideoxynucleotide capillary platforms, but are still new enough that questions of required sequencing depth of coverage and error models have yet to be conclusively defined. The HGSC is using both the Applied Biosystems (AB) SOLiD and Roche/454 platforms and is making significant contributions to each sector of the Project pilot. The three pilot project sectors are: Pilot 1 - low-depth of coverage sequencing of whole genomes for 180 individuals; Pilot 2 - deep sequencing of whole genomes for two parent-offspring trios; Pilot 3 - targeted sequencing of coding sequences for 1000 genes in ~1000 individuals.
Additional Resources
Learn more about the 1000 Genomes Project
BCM-HGSC and the International HapMap Project
BCM-HGSC is actively involved in the International HapMap Project.
This project is a partnership of scientists from around the world who are working to develop a distinctive map of the human genome that will help researchers find the genes associated with human diseases.
Common diseases, such as mental illness, epilepsy, diabetes, cancer, and heart disease, are affected by both DNA and environmental factors. Tiny variations in human DNA, called “single nucleotide polymorphisms” or SNPs, can influence how people differ in their risk for these diseases.
The HapMap project is developing a map of the common patterns of these DNA variations, in the hope that identifying them can lead to an understanding of the complex causes of disease in humans. This map will be a key resource for researchers to use when searching for genes that affect our health and our body’s responses to drugs and the environment.
As part of the International HapMap Project, BCM-HGSC is resequencing 2.5 million bases (Mb) of the “ENCODE” region of the human genome in order to identify novel SNPs. This 2.5 Mb target is actually five separate, equal-size segments located on chromosomes 7p15, 8q24, 9q34, 12q12, and 18q12. These five segments contain a number of candidate disease genes associated with hypertension, diabetes, and bone disorders.
For this project, we sequenced DNA samples from 48 individuals who are from four different regions of the world, including the United States, China, Japan, and Nigeria. All novel SNPs will be genotyped in 270 samples from these same four populations.
HapMap 3 and ENCODE 3 (Janary 2009 draft release 3)
Current data download
HapMap3 draft release 3 FTP data
Previous releases
HapMap3 draft release 2 FTP data
HapMap3 draft release 1 FTP data
About the Project
This is draft release 3 for genome-wide SNP genotyping and targeted sequencing in DNA samples from a variety of human populations (sometimes referred to as the "HapMap 3" samples).
This release contains the following data:
-
SNP genotype data generated from 1184 samples, collected using two platforms: the Illumina Human1M (by the Wellcome Trust Sanger Institute) and the Affymetrix SNP 6.0 (by the Broad Institute). Data from the two platforms have been merged for this release.
-
PCR-based resequencing data (by Baylor College of Medicine Human Genome Sequencing Center) across ten 100-kb regions (collectively referred to as "ENCODE 3") in 712 samples.
Since this is a draft release, we ask you to check this site regularly for updates and new releases.
Data Production Institutions
Funding Agencies
-
National Institutes of Health -- National Human Genome Research Institute (NHGRI)
-
National Institute on Deafness and Other Communication Disorders (NIDCD)
HapMap 3 Samples
The HapMap 3 sample collection comprises 1,301 samples (including the original 270 samples used in Phase I and II of the International HapMap Project) from 11 populations, listed below alphabetically by their 3-letter labels. For more information about these samples, click here.
| label | population sample | number of samples |
|---|---|---|
| ASW | African ancestry in Southwest USA | 90 |
| CEU | Utah residents with Northern and Western European ancestry from the CEPH collection | 180 |
| CHB | Han Chinese in Beijing, China | 90 |
| CHD | Chinese in Metropolitan Denver, Colorado | 100 |
| GIH | Gujarati Indians in Houston, Texas | 100 |
| JPT | Japanese in Tokyo, Japan | 91 |
| LWK | Luhya in Webuye, Kenya | 100 |
| MEX | Mexican ancestry in Los Angeles, California | 90 |
| MKK | Maasai in Kinyawa, Kenya | 180 |
| TSI | Toscans in Italy | 100 |
| YRI | Yoruba in Ibadan, Nigeria | 180 |
ENCODE 3 Regions
Five of the ten ENCODE 3 regions overlap with the HapMap-ENCODE regions; the other five are regions selected at random from the ENCODE target regions (excluding the 10 HapMap-ENCODE regions). All ENCODE 3 regions are 100-kb in size, and are centered within each respective ENCODE region. Read more about the ENCODE project here.
| region | chromosome | coordinates (NCBI build 36) | status |
|---|---|---|---|
| ENm010 | 7 | 27,124,046-27,224,045 | HapMap-ENCODE |
| ENr321 | 8 | 119,082,221-119,182,220 | HapMap-ENCODE |
| ENr232 | 9 | 130,925,123-131,025,122 | HapMap-ENCODE |
| ENr123 | 12 | 38,826,477-38,926,476 | HapMap-ENCODE |
| ENr213 | 18 | 23,919,232-24,019,231 | HapMap-ENCODE |
| ENr331 | 2 | 220,185,590-220,285,589 | New |
| ENr221 | 5 | 56,071,007-56,171,006 | New |
| ENr233 | 15 | 41,720,089-41,820,088 | New |
| ENr313 | 16 | 61,033,950-61,133,949 | New |
| ENr133 | 21 | 39,444,467-39,544,466 | New |
Data Content of This Release
A. SNP Genotype Data
| label | number of samples | number of QC+ SNPs | number of polymorphic QC+ SNPs |
|---|---|---|---|
| ASW | 71 | 1,632,186 | 1,536,247 |
| CEU | 162 | 1,634,020 | 1,403,896 |
| CHB | 82 | 1,637,672 | 1,311,113 |
| CHD | 70 | 1,619,203 | 1,270,600 |
| GIH | 83 | 1,631,060 | 1,391,578 |
| JPT | 82 | 1,637,610 | 1,272,736 |
| LWK | 83 | 1,631,688 | 1,507,520 |
| MEX | 71 | 1,614,892 | 1,430,334 |
| MKK | 171 | 1,621,427 | 1,525,239 |
| TSI | 77 | 1,629,957 | 1,393,925 |
| YRI | 163 | 1,634,666 | 1,484,416 |
| consensus | 1,115 | 1,525,445 | 1,490,422 |
B. PCR Resequencing Data
| label | number of samples |
|---|---|
| ASW | 55 |
| CEU | 119 |
| CHB | 90 |
| CHD | 30 |
| GIH | 60 |
| JPT | 97 |
| LWK | 60 |
| MEX | 27 |
| MKK | 0 |
| TSI | 60 |
| YRI | 120 |
| total | 712 |
Quality Control of This Release
A. SNP Genotype Data
Genotyping concordance between the two platforms was 0.9931 (computed over 249,889 overlapping SNPs). Data from the two platforms was merged using PLINK (--merge-mode 1), keeping only genotype calls if there is consensus between non-missing genotype calls (that is, merged genotype is set to missing if the two platforms give different, non-missing calls).
Quality control at the individual level was performed separately by the two sites. Only individuals with genotype data on both platforms were kept in this release. The following criteria were used to keep SNPs in the QC+ data sets:
-
Hardy-Weinberg p>0.000001 (per population)
-
missingness <0.05 (per population)
-
<3 Mendel errors (per population; only applies to YRI, CEU, ASW, MEX, MKK)
-
SNP must have a rsID and map to a unique genomic location
The "consensus" data set contains data for 1115 individuals (558 males, 557 females; 924 founders and 191 non-founders), only keeping SNPs that passed QC in all populations (overall call rate is 0.998). The "consensus|polymorphic" data set has 35,023 monomorphic SNPs (across the entire data set) removed.
In all genotype files, alleles are expressed as being on the (+/fwd) strand of NCBI build 36.
B. PCR Resequencing Data
The sequence-based variant calls were generated by tiling with PCR primer sets spaced approximately 800 bases apart across the ENCODE 3 regions. Following filtering low-quality reads the data were analyzed with SNP Detector version 3, for polymorphic site discovery and individual genotype calling. Various QC filters were then applied. Specifically, we filtered out PCR amplicons with too many SNPs, and SNPs with discordant allele calls in multiple amplicons. We also filtered out SNPs with low completeness in samples, or with too many conflicting genotype calls in two different strands.
In the QC+ data set, we filtered out samples with low completeness, and filtered out SNPs with low call rate in each population (<80%) and not in HWE (p<0.001). In the QC+ data set, the overall false positive rate is ~3.2%, based on a limited number of validation assays.
Caveats in This Release
A. SNP Genotype Data
-
Missing from this release are Illumina SNPs that are A/T or C/G due to strandedness issues.
-
Missing from this release are Illumina SNPs that are mitochondrial (as they do not have rsIDs).
-
There may be few remaining SNPs (Illumina) in this release that are still on (-/rev) strand of NCBI build 36, but they are not A/T or C/G SNPs, so easy to identify downstream.
B. PCR Resequencing Data
All variant calls have not yet been validated: we estimate that there is currently a false positive rate of ~12% among all calls, with a slightly higher rate (~14%) if considering just the singletons. Additional validation is ongoing. PCR sequencing of additional samples (MKK) is also ongoing.
How to Download This Release
A. SNP Genotype Data
To download the HapMap 3 data from our ftp site, click here.
B. PCR Resequencing Data
To download the ENCODE 3 data from our ftp site, click here.
Analysis Plans
Listed below are the analysis plans that we are currently pursuing:
-
SNP allele frequency estimation
-
Population differentiation
-
Linkage disequilibrium analysis
-
SNP tagging
-
Imputation efficiency
-
Genomic locations of human CNVs
-
Genotypes for CNVs
-
Population genetic properties of CNVs (allele frequencies, population differentiation, etc.)
-
Mutation rate (frequency of de novo CNV) and potential mutational mechanisms
-
Linkage disequilibrium properties of CNVs
-
Tagging and imputation of CNVs
-
Signals of selection around CNVs
-
Association of SNPs and CNVs with expression phenotypes
Data Release Policy
The release of pre-publication data from large resource-generating scientific projects was the subject of a meeting held in January 2003, the "Fort Lauderdale" meeting. An NHGRI policy statement based on the outcome of the meeting is on the NHGRI web site (https://www.genome.gov/10506537/reaffirmation-and-extension-of-nhgri-rapid-data-release-policies).
The recommendations of the Fort Lauderdale meeting address the roles and responsibilities of data producers, data users, and funders of "community resource projects", with the aim of establishing and maintaining an appropriate balance between the interests of data users in rapid access to data and the needs of data producers to receive recognition for their work. The conclusion of the attendees at the meeting was that responsible use of the data is necessary to ensure that first-rate data producers will continue to participate in such projects and produce and quickly release valuable large-scale data sets. "Responsible use" was defined as allowing the data producers to have the opportunity to publish the initial global analyses of the data, as articulated at the outset of the project. Doing so also will ensure that the data generated are fully described.
Mirror Sites